Sommaire
1.1-
Principe et généralités de la photocatalyse
hétérogène
1.1.1-
Le
rayonnement lumineux
1.1.2-
Le
dioxyde de titane
1.1.3-
La
photocatalyse hétérogène
a-
Terminologie
b-
Modes
d’actions du TiO2
1.1.4-
Applications
1.2-
Facteurs influençant la photocatalyse hétérogène
1.2.1-
La
photolyse directe
1.2.2-
Influence
de l’oxygène dissous
1.2.3-
Influence
du pH
1.2.4-
Influence
de la concentration en catalyseur
1.2.5-
Influence
de la concentration initiale en polluant
1.2.6-
Influence
du flux lumineux
1.2.7-
Influence
de la température
1.2.8-
Influence
du champ quantique
1.2.9-
Influence
d’ions en solution
1.2.10-
Influence
de la méthode de préparation et de la cristallinité du catalyseur
1.3-
Les réacteurs photochimiques et les sources
lumineuses
1.3.1-
Les
réacteurs photochimiques
1.3.2-
Les
sources lumineuses
Partie
I : Le traitement de l’eau par photocatalyse
La figure 1 représente la quantité d’eau existante à la
surface de la Terre (eaux des océans, des lacs, des rivières, des glaciers, …)1 . Le petit cube représente à partie
correspondante à l’eau potable2. Ce petit cube correspond
approximativement à 9000 km3 d’eau potable par an. Ce schéma illustre très bien
l’importance de bien maîtriser le stockage et le traitement de l’eau.
Ainsi, il est estimé que3 :
-
1,2
milliard de personnes (un quart de la population mondiale) manque d’eau potable
-
1,4
milliard de personnes sont sans évacuation efficace d’eaux usées.
-

Plus de 80 pays (>40% de la population mondiale)
souffrent d’un manque d’eau.
Le traitement de l’eau est séparé en deux parties
distinctes pour réduire les problèmes environnementaux croissants : (a) le
traitement chimique de l’eau potable contaminée, de l’eau souterraine et de
surface ; (b) le traitement chimique des eaux usées contenant des toxiques
ou des composés non biodégradables.
Depuis 25 ans, la recherche sur le traitement de l’eau
s’est fortement intensifiée. Les contoles de
pollution sont plus rigoureux et la législation
devient de plus en plus strictement sur les normes de l’eau distribuée.
De nouvelles techniques de traitement ont été développées
tels que les Procédés d’Oxydation Avancés (POAs). Ces techniques sont des
alternatives très intéressantes pour la dégradation de polluants organique non
biodégradables. Elles sont beaucoup plus efficaces que les techniques
habituelles de floculations, précipitation, adsorption sur charbon activés ou
osmose inverse. Le traitement chimique par les POAs peut conduire à la
minéralisation complète des plluants en CO2,
et dans le cas de composés halogénés à la formation d’ions halogènes.
Les POAs sont basées sur la propriété de génération et
d’utilisation des radicaux hydroxyles comme oxydant primaire pour la
dégradation de polluants organiques. Les POAs, tels que les systèmes
U.V.-peroxyde, ozone ou processus photo-Fenton ont largement démontré leurs
efficacités dans l’oxydation de composés organiques. Trois autres POAs (la photocatalyse, la sonolyse et la
radiolyse gamma) ont émergé ces dernières années.
La radiolyse concerne l’ensemble des transformations
chimiques provoquées par l’interaction de rayonnements ionisants avec le milieu
qu’ils traversent. Ces rayonnements peuvent être de nature variable (électrons,
photons, neutrons, particules chargées lourdes), mais le plus couramment
utilisé est le rayonnement gamma émis soit par le 60Co (photons de 1,17 et 1,33
MeV), soit par le 137Cs (photons d’environ 660 keV)4. Ces photons ionisent le solvant (aqueux
ou éthanolique), en produisant très rapidement
(quelques dizaines de nanosecondes) des espèces radicalaires :
![]() (1)
(1)
En présence d’air, les atomes d’hydrogène et les électrons
sont attirés par l’oxygène pour former des radicaux peroxyles
HO2°, O2°- :
![]() (2)
(2)
![]() (3)
(3)
![]() (4)
(4)
Le soluté dissous dans le solvant ainsi irradié ne subit
pas directement l’effet des rayonnements ionisants car sa concentration est
choisie suffisamment faible (en général inférieur à 10-3 M/L). pour que cet effet direct soit négligeable. En revanche, le
soluté dissous subit l’action des radicaux libres produits par la radiolyse du
solvant.
La radiolyse gamma est surtout utilisée pour étudier
l’oxydation de molécules d’intérêt biologique5, qu’il s’agisse de
glucides6, des protéines7, des acides nucléiques8,9, ou des lipides10. Une telle approche
permet en effet d’étudier les sites d’attaque de ces composés par des espèces
radicalaires produites par radiolyse, et de comprendre les mécanismes
réactionnels qui en résultent.
La sonolyse est une technique
nouvelle utilisant les ultrasons pour dégrader des polluants en milieu aqueux.
La propagation d’ultrasons dans l’eau conduit à la formation de micros bulles de cavitation. A l’intérieur de ces micros bulles règne des conditions extrêmes de température et de
pression qui conduisent à la dissociation de l’eau et à la production de
radicaux OH°, HOO°, etc. Il a été mis en évidence que des fréquences
supérieures à 400 kHz sont plus favorable à la formation de radicaux OH°.
Le concept de la dégradation photocatalytique
est simple, un semi conducteur solide stable est irradié pour stimuler des réaction à l’interface solide/liquide. En principe, le
semi-conducteur peut être réutilisé après plusieurs utilisations. L’équation
générale de la photocatalyse est la suivantes :
![]() (5)
(5)
Le trou h+ formé peut réagir avec un groupement hydroxyle
adsorbé à la surface du semi-conducteur pour former des radicaux OH° très
oxydants.
1.1-
Principe et généralité
de la photocatalyse
1.1.1-
Les lois
d’absorption de la lumière
La loi fondamentale de la photochimie, connue sous le nom
de loi de Grotthus-Draper, spécifie que seule la
radiation absorbée par un système peut initier une réaction photochimique11.
Si un flux d’intensité (I0) pénètre dans un
milieu homogène, il sera partiellement réfléchi à chacune des interfaces (IR)
et absorbé par le milieu (IA). L’intensité du flux lumineux
émergeant du milieu traversé sera en conséquence donnée par :
IT = I0
– IA – IR (6)
La description phénoménologique de l’absorption de la
lumière est donnée par une évaluation quantitative de l’atténuation de
l’intensité I dans la direction x d’un faisceau lumineux parallèle, perpandiculairement incident à la surface plane d’un milieu
transparent. La loi de Lambert-Bouguer est :
![]() (7)
(7)
![]() (8)
(8)
La constante d’absorption a (en cm-1) est un paramètre dépendant de la
longueur d’onde et est défini pour un solide isotrope. Elle est reliée au
coefficient d’absorption k par la relation :
![]() (9)
(9)
où
l0 est la longueur d’onde de la radiation dans le vide. Le
coefficient k représente la partie imaginaire de l’indice de réfraction
complexe du milieu, une grandeur caractéristique directement liée à sa
polarisabilité électrique et nécessaire au calcul de ses propriétés
optiques :
![]() (10)
(10)
 (11)
(11)
L’absorption optique d’une substance en solution peut être
obtenue par la loi de Lambert-Beer :
![]() (12)
(12)
Cette loi n’est strictement valable que pour des solutions
diluées. L’absorbance A (ou densité optique) est la quantité mesurée commune à
toutes les techniques photométriques. Elle dépend de la longueur du chemin
optique l (en cm) dans la solution, de la longueur d’onde et du coefficient de proportionnalité e.
La relation A=f(l) n’est autre que le spectre d’absorption. Le coefficient
d’extinction (ou d’absorption) molaire décadique e (en mol-1/L/cm) est une constante indépendante
de la concentration à une longueur d’onde déterminée et est directement relié à
la section efficace s
d’absorption de la lumière par les molécules en solution :
![]() (13)
(13)
De part leur définition, l’absorbance et la transmittance de plusieurs systèmes placés dans le trajet
optique sont des grandeurs additives et multiplicatives, respectivement.
Un système modèle simple, donné par une solution 10-3M
d’une substance caractérisée par un coefficient d’extinction de 104
L.mol-1.cm-1, montre qu’une couche aussi mince que l=2 mm absorbe 99% de la
lumière incidence (T=0,01 et A=2,0). Des transformations photochimiques ne
pourront donc avoir lieu que très près de la surface d’une telle solution. Pour
une application technique de synthèse photo-induite,
les conséquences chimiques de ce phénomène dépendront du procédé de réaction
(réacteur à film mince, mélange efficace, concentrations les plus faibles, …).
Le calcul des conditions d’absorption dans un réacteur en opération est rendu
encore plus difficile par la poly-chromaticité des sources lumineuses, de
géométries d’irradiation complexes, de mélanges de substances absorbantes, etc…
Le traitement quantitatif de l’absorption de la lumière
n’est pas limité au seul cas des milieux continus (transparents). Les systèmes
hétérogènes constituent en fait la majorité des cas rencontrés dans les
applications les plus importantes de la photochimie.
Sur une surface uniforme plane, dite spéculaire
(miroir) ; l’absorption et la réflexion de la lumière sont reliées par les
lois de Fresnel :
![]() (14)
(14)
![]() à
incidence normale j=0 (15)
à
incidence normale j=0 (15)
La valeur de la réflectance
spéculaire augmente rapidement avec le coefficient d’absorption (typiquement de
5% pour k=0 à pratiquement l’unité pour k>10, soit a>106 cm-1 dans le domaine du visible). Plus
le coefficient d’absorption est élevé et plus la réflexion spéculaire joue donc
un rôle important en perturbant l’absorption de la lumière par le solide.
Dans le cas d’un milieu constitué de nombreuses interfaces
et dans lequel les dimensions des particules sont suffisantes pour que les manifestations
de la réflexion, de la réfraction et de la diffraction restent définies,
l’orientation dans toutes les directions possibles des faces d’une phase
dispersée provoquera la réflexion de la lumière incidente selon tous les angles
appartenant à l’hémisphère d’où elle provient. Dans de telles conditions, où la
distribution angulaire de la radiation réfléchie ne dépend pas de l’angle
d’incidence, on parle de réflexion diffuse.
La première loi de la réflexion diffuse a été proposée par
Lambert sur la base de l’observation qu’une surface blanche illuminée par le
soleil possède le même éclat à tous les angles d’observation :
![]() (16)
(16)
Le flux de radiation IR réémise par unité de
surface z et par unité d’angle solide w est donc proportionnel au cosinus des angles d’incidence q et d’observation u. La constante A, appelée albédo, donne la fraction de
l’énergie réfléchie, et est en conséquence toujours inférieur à 1. Le
coefficient B est appelé la densité de radiation d’une surface (en W/m²/radian).
Cette loi, dite du cosinus de Lambert est rigoureusement valable pour un
radiateur idéal.
1.1.2-
Le
dioxyde de titane
L’oxyde de titane est un semi-conducteur qui existe sous
différentes formes cristallographiques12. L’oxyde le plus faible est
TiO. Il appartient à la grande famille des oxydes TinO2n-1
qui sont décrit comme ayant une structure cristalline déformée de type CS13,
et qui incluse la phase Magnéli (4 £ n £
9). Le plus grand oxyde est TiO2, qui existe sous différentes formes
cristallines : le rutile, l’anatase, la brookite,
et un grand nombre de phases obtenues sous hautes pressions. Pour ajouter à
cette complexité, le TiO2 peut accepter les formes non
stœchiométriques TiO2-x.
Le rutile a un réseau tétraédrique de cations. Des
octaèdres d’oxygène entourent les cations. L’octaèdre TiO6 partage
une arrête commune le long de l’axe [001] et un sommet commun avec un autre
octaèdre adjacent, avec un arrangement de contact cation – anion - cation. Bien
que le rutile soit un isolant, par l’ajout de petites quantités de Ti3+,
la conductivité électrique peut être induite via des interactions cation - cation ou Ti3+ - anion -
Ti4+ 14. La distance inter-ionique moyenne15
dans le rutile est de 1,959 A pour la liaison Ti-O
ainsi que 2,96 A et 3,57 A pour les liaisons Ti-Ti.

L’anatase est une structure tétraédrique allongée16
avec des octaèdres d’oxygène irrégulier, mais les distances Ti-O
(1,917 A de moyenne) sont sensiblement égales aux autres côtés et à celles du
rutile.

La brookite17 est orthorhombique avec une
structure plus complexe, bien que les distances Ti-O
soient similaires aux autres structures.

Les phases hautes pressions ont une structure columbite (a-PbO2), avec des octaèdres TiO6 qui
partagent deux arrêtes, les arrêtes partagées ont des distances O-O plus
petites.

|
|
Anatase |
Rutile |
Brookite |
Phases hautes pressions |
|
Ti-O (en A) |
1.917 |
1,959 |
|
|
|
Ti-Ti (en A) |
|
2,96 |
|
|
1.1.3- La
photocatalyse hétérogène
a-
Terminologie
Le terme de photocatalyse est encore sujet à beaucoup de
débat. Par exemple, Suppan18 est coll. affirme que l’idée de
réaction photocatalytique est fondamentalement incorrecte, en expliquant que
dans une réaction, la lumière est suppléant au catalyseur, lequel est toujours
le réactant principal.
En réalité, le terme de photocatalyse est plus vaste, il ne
repose pas sur une action catalytique de la lumière, mais plutôt sur une
accélération de la phototréaction par la présence du
catalyseur. Le terme de photoréaction est parfois
remplacé par réaction photoinduite ou par réaction
photoactivée19.
La définition la plus correcte de la photocatalyse incluse
le processus de photosensibilisation, par exemple, par lequel une altération
photochimique est réalisée sur une espèce chimique résultante de l’absorption
initiale de radiation par d’autres espèces chimiques appelées photosensibilisateurs.
La photocatalyse hétérogène implique des photoréactions qui se produise à
la surface du catalyseur. Si le processus de photoexcitation
initiale se produit sur une molécule adsorbée, laquelle alors interagi avec le
catalyseur, le processus est appelé photoréaction
catalysée. Si la photoexcitation initiale lieu sur le catalyseur et qu’ensuite le
catalyseur photoexcité réagi alors avec une molécule
adsorbée, on parle de photoréaction sensibilisée. Dans tous les cas, la
photocatalyse hétérogène fait référence à un semi-conducteur photocatalyseur ou
à un semi-conducteur photosensibilisateur.
Les recherches sur la photocatalyse ont beaucoup progresser
durant les années 70 concernant le comportement photoélectrochimique
des semi-conducteurs d’oxydes métalliques à large bande gap grâce aux travaux
de Fujishima et Honda20. Ces deux auteurs
se sont interessés à la dissociation de l’eau photoinduite par
des électrodes de TiO2 rutile.
Malgré le fort engouement dans ces recherches, la
conversion efficace en hydrogène par le soleil n’a pas dépasser
quelques pour-cent à cause de la faible quantité de lumière recueillie par le
TiO2, l’absorption fondamentale se situe dans la région des U.V.
(figure 3).

Figure 3 : comparaison du spectre solaire et du
spectre d’absorption du TiO2
Dans le milieu des années 80, l’intérêt s’est porté sur
d’autres applications des oxydes métalliques semi-conducteur. La recherche
s’est tournée vers la destruction de polluants par action photocatalytique du
TiO2 dans l’eau.
b-
Mode d’action du TiO2
Deux formes cristallines du TiO2 ont une
activité photocatalytique, l’anatase et le rutile21. L’anatase a une
bande gap de 3,23 eV (384 nm) et le rutile de 3,02 eV (411 nm)22. L’anatase
a été montré comme étant la forme le plus active. Le spectre d’action pour
l’anatase montre une diminution très rapide dans l’activité après 385 nm
(figure 3).
Le processus photocatalytique repose sur l’excitation du
TiO2 par un rayonnement lumineux de longueur d’onde inférieur à 400
nm (figure 4). Un électron passe de la bande de valence à la bande de
conduction, créant un site d’oxydation (un trou h+) et un site de
réduction (un électron e-).
![]() (17)
(17)
Les indices BC, BV et ads
signifient respectivement bande de conduction, bande de valence et adsorbé.

Figure 4 : schéma du mécanisme de dégradation
photocatalytique
Les trous h+ réagissent avec les donneurs
d’électrons tels que l’eau, les anions OH- et les produits organiques
R adsorbés à la surface du semi-conducteur (équations 6 à 8) en formant les
radicaux hydroxyles et R° :
![]() (18)
(18)
![]() (19)
(19)
![]() (20)
(20)
Les électrons réagissent avec des accepteurs d’électrons
tels que le dioxygène pour former des radicaux
superoxydes23. Cette réaction limite la recombinaison des charges
(équation 9) :
![]() (21)
(21)
En l’absence d’accepteurs et de donneurs d’électrons
appropriés, on assiste à l’annihilation trou/électron (réaction de
recombinaison très rapide de l’ordre de la picoseconde) :
![]() (22)
(22)
Cette dernière réaction explique l’importance de l’eau et
de l’oxygène dans la réaction de dégradation photochimique24. La recombinaison
trou/électron est un facteur qui limite l’efficacité de cette méthode car la
probabilité de recombinaison est d’environ 99,9%.
De plus, seule une fraction du spectre solaire (5% environ,
figure 3) est effectivement utilisée pour la dégradation. Il existe plusieurs
solutions pour pallier cet inconvénient : le dopage du semi-conducteur par
d’autres métaux25 (pour élargir la gamme d’absorption du rayonnement
U.V.) ou encore l’addition au milieu réactionnel d’accepteurs d’électrons
(ozone, peroxyde d’hydrogène, Fe3+, …)26.
![]() (23)
(23)
![]() (24)
(24)
![]() (25)
(25)
Le peroxyde d’hydrogène a le double avantage d’absorber
dans l’U.V. et de conduire à la formation de nouveaux radicaux hydroxyles :
![]() (26)
(26)
Malheureusement, le coefficient d’absorption molaire est
faible entre 300 et 400 nm (composante U.V. du spectre lumineux de la lampe).
Il est alors nécessaire d’utiliser une très forte concentration en peroxyde
d’hydrogène pour provoquer l’oxydation efficace des produits.
1.1.4-
Applications de la photocatalyse à la décontamination de l’eau
Le dioxyde de titane est un des matériaux les plus basiques
dans notre vie de tout les jours. Il a été utilisé
dans une grande variété de peintures, de cosmétiques et dans l’alimentaire.
Aujourd’hui, la consommation annuelle de TiO2 dans le monde dépasse
les trois millions de tonnes.
Naturellement, le type de TiO2 qui est utilisé
comme pigment est différent de celui utilisé en photocatalyse. La photoactivité du dioxyde de titane conduit à la
décomposition des molécules organiques qui viennent à sa surface. Ce phénomène
peut être un problème, car les peintures contiennent des molécules qui peuvent
être dégrader par l’action photocatalytique.
Dans d’autres cas, la photoactivité
du TiO2 joue un rôle positif : par exemple pour la dégradation
de produits chimiques malodorants ou irritant, pour des produits toxiques, des
bactéries, etc… Pour toutes ces raisons, la
technologie photocatalytique est promis à devenir une
nouvelle importante industrie.
Quand le TiO2 capture un rayonnement UV, il se forme de
l’oxygène activé à partir d’eau et d’oxygène de l’air à la surface du
catalyseur. Ce procédé est similaire à la photosynthèse, dans laquelle la
chlorophylle capture, sous lumière solaire, de l’eau et du dioxyde de carbone
pour donner de l’oxygène et du glucose. L’oxygène activé formé est fortement
oxydant et décompose les molécules organiques et tue les bactéries. Récemment,
l’industrie du bâtiment a utilisé la photocatalyse sur couche mince pour ces
propriétés stérilisantes, déodorantes et anti-salissantes.
Actuellement, le phénomène super hydrophile sur le
photocatalyseur a été mis en évidence. Dans notre environnement quotidien, la
surface d’un matériau repousse l’eau de quelques degrés. Ce degré d’eau
repoussé est mesuré à l’aide d’une goutte d’eau posée à la surface du matériau.
Sur le verre ou d’autres matériaux inorganiques, l’eau à un angle de contact
compris en 20 et 30 degrés. Avec des résines, l’angle de contact de l’eau est
généralement compris entre 70 à 90 degrés. Avec une résine hydrophobique, telle
que une résine siliconée, l’angle de contact est supérieur à 90 degrés.
Peu de matériaux ont un angle de contact inférieur à 10 degrés,
avec comme exception quelques matériaux adsorbant l’eau et qui ont été activées
par du savon ou d’autres agents similaires. Lorsque l’angle de contact devient
proche de zéro, la surface de matériau ne retient plus l’eau et on dit qu’elle
est « super-hydrophile ». Cependant, ces surfaces n’ont pas de
caractère hydrophile très longtemps.
Quand la surface photocatalytique est exposée à un
rayonnement UV, l’angle de contact à la surface du TiO2 avec l’eau
diminue graduellement. Après une exposition suffisante à la lumière, la surface
devient super hydrophile. Ce caractère particulier peut être obtenue pendant un
ou deux jours. L’angle de contact augmente progressivement durant cette périodes et devient hydrophobe. A ce stade, la super hydrophilité peut être retrouvé grâce à une exposition à la
lumière UV. Ce type de photocatalyseur est le seul connue
possédant une propriété super hydrophile semi-permanente.
La figure 5 représente une utilisation du caractère super
hydrophile appliqué à une plaque de verre. Seule la partie droite du verre a
été irradié et est super hydrophile. Lorsque de la vapeur d’eau est projetée
sur la plaque, la partie de droite ne retient absolument pas l’eau. La partie
droite reste totalement transparente.
Bien qu’une surface super hydrophile est
très utile, dans certains cas une surface super hydrophobe est préférable27.
Par exemple, sur les pare-brises automobiles, sur une surface super hydrophile,
le vent créé une distorsion de l’eau les jours de pluies. Pour une telle
applications, une surface super hydrophobe, c’est à dire ayant un angle de
contact supérieur à 150°, permet d’éliminer immédiatement l’eau. La surface
super hydrophobe est obtenue par dépôt en film mince d’un gel de boehmite (AlOOH) et d’acetylacetonate (Al(C5H7O2)3). Le
film est ensuite calciné à 500°C. Le film le plus super hydrophobe obtenue par cette méthode possède un angle de contact de
160° avec l’eau. Généralement, le caractère super hydrophobe se détériore
rapidement à l’extérieur car la surface se sature en matières grasses. C’est à
ce moment que le TiO2 joue un rôle très important. En effet, l’incorporation de
TiO2 dans le gel permet d’obtenir une photoactivitée
suffisante pour nettoyer la surface et augmenter la durée de vie de l’effet
super hydrophobe. L’ajout de 0,2% de TiO2 permet de garder une surface
transparente et d’augmenter la durée de vie de 1000h à l’exposition extérieure.
La figure ci-dessous représente une goutte d’eau posée sur une surface super
hydrophobe.

1.2-
Facteurs influençant la
photocatalyse hétérogène
1.2.1-
La
photolyse directe
La dégradation photochimique d’une molécule nécessite
l’absorption d’une radiation de longueur d’onde appropriée. Lors de
l’absorption des photons, les molécules passent à un état excité et cet excès
d’énergie peut être dissipé par plusieurs voies de désactivation qui
entraîne :
-
des
modifications physiques : processus de désactivation radiatifs
(fluorescence) ou non radiatif (transfert d’énergie à une molécule ou
photosensibilisation, transition d’un excité à un autre état d’énergie
inférieure).
-
Des
modifications chimiques : la molécule peut subir une modification directe
de son état excité en produit stable ou conduire à un intermédiaire réactif par
exemple un radical, capable d’amorcer une réaction en chaîne. Ces modification peuvent avoir lieu sur la molécule isolée
ou faire intervenir un autre réactif, par exemple l’oxygène.
L’absorption d’un quantum
d’énergie ne peut exciter qu’une seule molécule. Le rendement quantique f d’une réaction photochimique est défini par le rapport
entre le nombre de molécules qui réagissent et le nombre de photons absorbés en
un temps donné.
 (27)
(27)
avec r : le nombre de moles
réagissant par unité de temps,
Ia : le nombre de moles de photons absorbés
par unité de temps,
Trois cas sont envisageable,
-
si f = 1 : chaque photons absorbé produit une
transformation chimique,
-
si f < 1 : d’autres processus de désactivation entrent
en compétition avec la transformation chimique,
-
si f > 1 : une réaction en chaîne se met en place à
partir d’un produit de transformation chimique.
Le domaine de longueur d’onde utilisé se situe généralement
entre 200 et 700 nm (U.V. et visible). Les radiations
dans le proche infrarouge sont trop peu énergétique pour amorcer es réactions
chimiques.
Cependant, la
photodégradation est très lente. Par exemple, il faut dix jours pour diminuer
de 50% une solution d’acrinathrin à 50 mg/L. Une
solution de 100 mg/L de pentachlorophénol, à pH 7,3,
est décomposée de moitié en 48 heures.
Bien que les vitesses de réaction photolytique soit différentes d’une solution à l’autre, Tomin28
a classé une centaine de substances par demi-vie photolytique.
1.2.2-
Influence
de l’oxygène dissous
Dans la purification de l’eau par photocatalyse hétérogène,
les polluants sont généralement organiques. L’équation de la réaction en
présence d’oxygène peut s’écrire :
polluants organiques + O2
à CO2 + H2O
+ acides minéraux (28)
La littérature fournie un large regard sur l’influence de
l’oxygène. L’oxygène est nécessaire pour la minéralisation complète et ne doit
pas être en compétition d’adsorption avec d’autres espèces réactives sur le TiO2.
L’oxygène limite la recombinaison trou électron et forme des espèces O2°-
très réactives.
La concentration en oxygène affecte directement la vitesse
de réaction, qui est plus rapide quand la pression en oxygène (pO2)
dans l’eau augmente. Dans tous les cas, la différence entre de l’air (pO2
= 0,21 atm) et de l’oxygène pur (pO2 = 1 atm) n’est pas drastique (figure 1). Ceci est très
encourageant dans le cas d’un pilote industriel car l'utilisation de l’oxygène
pur entraîne un coût non négligeable.

Figure 1 : Effet de la
concentration en oxygène dissous sur la minéralisation photocatalytique
La figure 7 représente l’effet de l’oxygène sur la
cinétique de dégradation d’un polluant suivi par le TOC. Lorsque tout l’oxygène
a été consommé, la photodécomposition suivie par le TOC s’arrête. A ce moment,
de l’oxygène est injecté dans le milieu est la photodégration continue. Une
entré d’oxygène dans le réacteur n’est pas toujours nécessaire. En effet, l’eau
peut s’oxygéner par une vive agitation sous atmosphère.
L’ajout d’accepteur d’électron permet de réduire le
phénomène de recombinaison trou/électron. L’oxydant inorganique le plus étudié
pour son effet positif sur la cinétique de photodégradation est le peroxyde
d’hydrogène.
Le peroxyde d’hydrogène est meilleur accepteur d’électrons
que l’oxygène29. L’énergie minimum requise à la réduction de
l’oxygène pour réduire des radicaux hydroxyles est de 3,0 eV
tandis que pour le H2O2 elle est de 2,2 eV. De plus, l’eau oxygénée peut produire des radicaux
hydroxyles OH° par rupture photolytique dépendant de la longueur d’onde de la
radiation incidente. Cependant, il a été montré que l’effet de H2O2,
sur la cinétique de dégradation, n’est pas toujours positif et cela dépend du
système étudié. Le pH du milieu joue un rôle important car l’ajout de H2O2
conduit à la formation de radicaux hydroperoxyles HO2°
en milieu acide
![]() (29)
(29)
Le pKa de la réaction précédente
est de 4,88. La réaction est donc favorisée en milieu acide. Pour de grandes
concentrations en peroxyde d’hydrogène, les réactions suivantes se
produisent :
![]() (30)
(30)
![]() (31)
(31)
A partir de ces deux réactions, on voit bien qu’un grande concentration en H2O2 à
un effet négatif sur la cinétique de photodégradation. En effet, ces deux
réactions consomment les radicaux hydroxyles et hydroperoxyles
nécessaire à la photodagradation des molécules
organiques.
1.2.3-
Influence
du pH
Le pH en solution aqueuse affecte énormément le TiO2
sur sa charge de surface et la taille des agrégats. Le pH pour lequel la charge
de surface de l’oxyde est nulle s’appelle Point de Zéro Charge (pHPZC),
il est 6,5 environ pour le TiO2. Avant et après ce pH, la surface de
l’oxyde est chargée :
TiOH2+ à TiOH +
H+ pH
< 6.5 (32)
![]() (33)
(33)
TiOH
à TiO- + H+ pH > 6.5 (34)
![]() (35)
(35)
Les constantes d’équilibre de ces réactions ont été
déterminées par Kormann30 et al., pKTiOH2+
= 2,4 et pKTiOH = 8. La
spéciation des espèces en fonction du pH est la suivante :
TiOH ≥ 80% 3<pH<10 (36)
TiO- ≥ 20% pH>10 (37)
TiOH2+ ≥ 20% pH<3 (38)
Dans ces conditions, la dégradation photocatalytique de
composés organiques ionisés est très affectée par le pH. A première vue, pour
un polluant qui possède un pKa en dehors de la gamme 1-13, une solution très
acide semble être préjudiciable et une solution très basique semble être
favorable. La variation est très faible ou inexistante pour une solution
neutre.
L’influence du pH sur la taille des particules de TiO2 en
suspension aqueuse est représentée sur la figure 2.

Figure 2 : Influence du pH
sur la taille moyenne des particules de TiO2 P25
en solution aqueuse ([TiO2]=0,2 g/L)
Lorsque le pH approche du pHPZC, la charge de surface de
l’oxyde disparaît. Il y a donc beaucoup moins d’interactions électrostatiques
qui favorise la dispersion du catalyseur en suspension. Cela induit un
phénomène d’agrégation et de formation de cluster de TiO2. Il est donc normal
d’obtenir une baisse de la réactivité photocatalytique à pH 7 car les clusters
limite la transmission et l’absorption de la lumière. De plus, de larges
clusters sédimentent plus facilement que de très faibles particules, ce qui
nécessite une agitation plus vigoureuse pour maintenir une solution
relativement homogène. Par contre, cette variation de la taille des particules
peut être un avantage pour la séparation
(par sédimentation ou filtration) du photocatalyseur de la solution traitée.
1.2.4-
Influence
de la concentration en catalyseur
Dans un photoréacteur statique ou dynamique, la vitesse de
réaction initiale est directement proportionnelle à la masse de catalyseur
engagée. Cela indique que le système catalytique est vraiment hétérogène.
Cependant, à partir d’une certaine valeur, la vitesse de réaction devient indépendante
de la masse en catalyseur. Cette limite dépend de la géométrie et des
conditions de travail du photoréacteur. En effet, pour une quantité bien
définie de TiO2, il est nécessaire de connaître la surface de
catalyseur effectivement irradiée. Quand la concentration en catalyseur est
très grande, la lumière atteint difficilement le cœur du réacteur.

Figure 3 : Différents
photoréacteurs de laboratoire et les zones de pénétration
lumineuse pour différents chemins optiques.
Un grand nombre de chercheurs se sont intéressés à
l’influence de la concentration en catalyseur sur l’efficacité du procédé.
Malgré des résultats différents, l’idée qui émerge est que la radiation
incidente dans le photoréacteur et le chemin optique sont
fondamentaux dans la détermination de la concentration optimale en
catalyseur :
- Si la
lampe est dans le réacteur, la concentration optimale en TiO2
est très grande (environ plusieurs grammes par litre de solution) si le
chemin optique est court (plusieurs mm). Dans les autres cas, plusieurs
centaines de mg sont nécessaire pour un chemin optique de quelques cm.
- Si la
lampe est à l’extérieur du réacteur, mais que le chemin optique est court
(1 à 2 cm), la vitesse maximale est obtenue avec une concentration en TiO2
de 1 à 2 g/L.
- Si la lampe
est à l’extérieur du réacteur, mais que le chemin optique est de plusieurs
centimètres, la concentration en catalyseur appropriée est de plusieurs
centaines de milligrammes par litre de solution.
Dans tous les cas décrit, un effet d’écran est produit
quand la concentration en TiO2 est très grande. La vitesse de
réaction diminue à cause d’une opacité de la solution qui empêche le catalyseur
d’être illuminé. De plus, la taille des particules joue également sur l’opacité
de la solution.
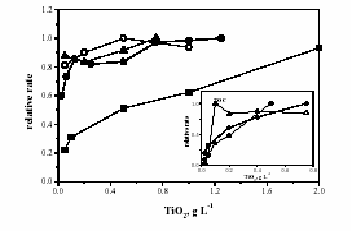
Figure 4 : Influence de la
concentration en catalyseur sur la cinétique de photocatalyse (les cinétiques
normalisées ont été utilisées) dans différents réacteurs. La ligne continue
sert à clarifier la tendance.
Pour bien caractériser un réacteur, il est faut déterminer
la quantité minimale de catalyseur pour laquelle la vitesse de réaction est la
plus grande. Mais, il n’est pas nécessaire de tester une grande gamme de
concentration. Généralement, la vitesse de réaction augmente très faiblement
avec la concentration en catalyseur, sauf aux faibles concentrations (inférieur
à 100 mg/L) où le phénomène est plus visible. Après que la vitesse de réaction
se soit stabilisée pour des concentrations plus élevées en TiO2, la
vitesse de réaction va diminuer. Alors, il n’est pas nécessaire d’augmenter la
concentration en catalyseur.
1.2.5-
Influence
de la concentration initiale du polluant
Beaucoup d’auteurs affirment que, pour une faible
variation, l’expression de la vitesse de photominéralisation des polluants
organiques suit la loi de Langmuir-Hinshelwood (L-H). Quatre cas ont alors
possible :
- La
réaction a lieu entre deux substances adsorbées
- La
réaction se produit entre un radical en solution et le polluant adsorbé
- La
réaction se produit entre un radical de la surface et le polluant en
solution
- La
réaction se produit entre les deux espèces en solution
Dans tous les cas, l’expression de l’équation est similaire
au modèle de L-H. Pour les études cinétiques seulement, il n’est pas possible
de déterminer si le processus a lieu à la surface du catalyseur ou en solution.
Bien que l’isotherme de L-H a été rapidement utilisé dans la modélisation, il
est généralement admis que la constante de vitesse et l’ordre sont
« apparents ». Ils servent à décrire la vitesse de dégradation, et
peuvent être utilisés pour optimiser un réacteur, mais ils n’ont pas de réalité
physique, et ne peuvent être utilisés pour identifier les réactions de surface.
Dans le traitement des données à partir du modèle de L-H,
il est admis que les réactions se produisent à la surface du catalyseur. Dans
ces conditions, deux situations extrêmes sont définies pour illustrer
l’adsorption à la surface du catalyseur : (i) le substrat et l’eau sont en
compétition d’adsorption sur les sites actifs du catalyseur et (ii) le réactant
et le solvant sont adsorbés à la surface sans compétition sur les mêmes sites
actifs du catalyseur.

Figure 5 : Courbes type de
cinétique de photodégradation.
En accord avec le modèle de L-H, la vitesse de réaction (r)
est proportionnelle à la fraction de surface recouverte par le substrat (qx). L’expression
obtenue est la suivante :
![]() (39)
(39)
![]() (40)
(40)
Où kr est la constante
de vitesse de la réaction, K est la constante d’adsorption du réactant, C est
la concentration au temps t, Ks est la constante d’adsorption du
solvant et Cs la concentration en solvant (dans l’eau Cs » 55,5 M). Comme Cs>>C et Cs pratiquement constant, la
partie recouverte par l’eau est toujours identique. En intégrant l’équation
précédente, on obtient :
![]() (41)
(41)
![]() (42)
(42)
Quand C0 est très faible, l’équation peut être
réduite en une équation de cinétique de première
ordre :
![]() (43)
(43)
Ainsi, si ln(C0/C) est
tracé en fonction du temps (ou de l’énergie accumulée), une droite est obtenue
qui permet de déterminer la constante de vitesse apparente (figure 3). De même,
aux fortes concentrations, les équations peuvent être simplifiées pour s’adapter
à l’ordre zéro. L’équation devient alors (C0-C) = krt comme représenté sur la figure 3.
En utilisant le modèle de L-H, des courbes similaires à la
figure 4 peuvent être obtenues à partir des données
expérimentales et par linéarisation des équations. L’effet de la concentration
initiale sur la vitesse de dégradation est représenté figure 4a. Le palier
obtenu pour de grande concentration initiale est du à une saturation de la
surface du catalyseur. La figure 4b représente la linéarisation qui permet de
déterminer la vitesse de dégradation kr. Finalement, les équation
4.7 sont obtenues à partir des équation 4.5 où la concentration est la moitié
de la concentration initiale (C/C0 = 0,5) :
![]() (44)
(44)
![]() (45)
(45)

Figure 6 : Données ajustées
selon le modèle cinétique de Figure
La photodécomposition conduit à des intermédiaires qui
peuvent s’adsorber à la surface du catalyseur. La concentration de ces
intermédiaires varie en fonction de la vitesse de leur minéralisation.
L’équation 4.4 devient alors :
 (46)
(46)
où
i est le nombre d’intermédiaires formés durant la dégradation (le solvant est
inclus dans la somme).
La connaissance des vitesses de réaction et comment la
vitesse de réaction est influencée par les différents paramètres est importante
pour la conception et l’optimisation de systèmes industriels. Les constantes de
vitesse de réaction de L-H sont utilisées pour comparer les vitesses de
réaction sous différentes conditions expérimentales. Une fois que les
constantes kr et K ont été évaluées, la
disparition du réactant peut être estimée si tous les facteurs restent
constants. Ainsi, une série de tests à différentes concentrations en substrat
ont été nécessaire pour démontrer que les résultats expérimentaux peuvent être
ajustés avec ce modèle. La gamme de concentration a été bien large pour
permettre la linéarisation de L-H. Cela entraîne, qu’il est possible de
déterminer la vitesse initiale aux faibles concentrations jusqu’à la limite ou
la relation entre la réaction initiale et la concentration initiale restent
constantes.

Figure 7 : Cinétique de
dégradation en fonction de la concentration initiale en polluant.
La figure 7 représente la vitesse initiale calculée. De 0,2
à 0,4 mM de substrat, la vitesse initiale est croissante. A cette
concentration, la saturation du catalyseur se produit et la vitesse de réaction
devient constante. La constante peut être calculée par le graphique dans la
figure 5 en utilisant le modèle de L-H.
![]() (47)
(47)
kr = 1,57mM/h
K
= 16,75 mM-1
Pour connaître kr et K, il est nécessaire de
dégrader le substrat à différentes concentrations C0. Ainsi, la
définition du volume du réacteur et de la surface sont
possible. Si l’énergie accumulée est utilisée, l’équation 3.24 permet d’obtenir
la constante de cinétique et parfois, la conception du réacteur.
1.2.6-
Influence
du flux lumineux
Depuis 1990, l’intérêt pour les technologies solaires qui
permettent la dépollution n’a cessé
d’augmenter. Les expériences initiales avec un réacteur parabolique permettant
la dépollution de l’eau et des fumés
pour le traitement en phase gazeuse, ont évoluées vers des systèmes à flux
faibles. L’utilisation d’un système solaire pour le traitement de l’eau est
très avantageuse car l’ordre de la vitesse de dégradation est peu influencé par
l’intensité lumineuse. Des expériences ont montré qu’au dessus d’un certain
flux photonique, l’influence de l’intensité sur la vitesse de réaction diminue
de l’ordre 1 à l’ordre 0,5 (Ollis 1991, Herrmann 1995). D’autres chercheurs, travaillant sous des
intensités d’irradiations particulières, n’ont pas obtenus les mêmes résultats,
mais probablement à cause des conditions expérimentales. D’autres auteurs
impute la transition r=f(I1) à r=f(I0,5)
à un excès d’espèces photogénérées (e-, h+ et OH°). Ceci
peut être démontré de la façon suivante, Les cinq équations de bases
sont :
TiO2 + hn à e- + h+ (48a)
e- + h+
à N + énergie (48b)
A + e- à A- (48c)
D
+ h+ à D+ (48d)
A-
+ D+ à intermédiaires à produits (48e)
L’étape limitant la réaction étant la réaction (16e). D’où,
![]() (49)
(49)
Pour un semi-conducteur de type n, comme le dioxyde de
titane, Les trous photo-induit sont moins nombreux que les électrons : [h+]<<[e-]. Les trous sont donc les espèces actives
limitantes. Ainsi,
![]() (50)
(50)
A tout instant, on a :
![]() (51)
(51)
ainsi,
![]() (52)
(52)
et
![]() (53)
(53)
A partir de l’équation du dessus, on peut voir que la
vitesse de réaction est directement proportionnelle au flux lumineux. Pour de
grand flux, la concentration instantanée en électrons et trous devient plus
importante que le terme kd[D]. de l’équation (20) :
![]() (54)
(54)
soit
![]() (55)
(55)
La vitesse de réaction devient :
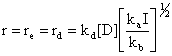 (56)
(56)
On retrouve bien I0,5
et que plus la vitesse de formation trou/électron est grande, plus la vitesse
de photocatalyse est importante. Il y a une utilisation optimale de la
puissance lumineuse qui correspond à la région où r est proportionnelle à I
(figure 8).

Figure 8 : Influence du flux
lumineux I sur la cinétique de photodégradation r
Pour de grandes intensités irradiantes, la transition de r
= f(I0,5) à r = f(I0) est
obtenue. A ce moment, la réaction photocatalytique n’est plus dépendante de la
radiation incidente, mais essentiellement du transfert de masse. Cet effet peut
être du à différentes causes, tels que le manque d’électrons pour O2
par exemple, Les molécules organiques à proximité de la surface du TiO2
et/ou un excès de produit occupant les sites actifs du catalyseur.
Actuellement, ce phénomène apparaît plus fréquemment dans les travaux avec du
catalyseur supporté, et/ou lorsque l’agitation est faible, ce qui implique une
plus petite surface en contact avec la solution. L’intensité à laquelle se
produit le changement d’ordre est différente suivant les conditions opérations
du système.
1.2.7-
Influence
de la température
Le système photocatalytique ne requiert pas de chaleur car
il s’agit d’un processus d’activation photonique. L’énergie d’activation vrai
Et est nulle, bien que l’énergie d’activation apparente Ea
soit très faible (quelques kJ/mol) pour une gamme de température de 20-80°C.
Cependant, à très faible température (-40-0°C), l’activité diminue et l’énergie
d’activation Ea devient positive. A
l’inverse, aux « grandes » températures (70-80°C) pour différents
types de réactions photocatalytiques, l’activité diminue et l’énergie
d’activation Ea devient négative. Ce
comportement peut être expliqué à partir des mécanismes de
Langmuir-Hinshelwood. La diminution de la température favorise l’adsorption,
qui est un phénomène spontanément exothermique. Le paramètre q de l’équation (7) tend vers l’unité, donc KC devient
>>1. De plus, la faible température favorise également l’adsorption des
produits de réaction. Au contraire, quand la température augmente au dessus de
80°C, proche du point d’ébullition de l’eau, l’adsorption exothermique des
polluants est défavorisée.
D’autres conséquences industrielles peuvent être
considérées. Si la température est grande, les matériaux utilisés pour les
installations doivent être résistants aux chaleurs (dilatable), et la
concentration en oxygène dans l’eau va diminuer. En conséquence, la température
optimale est généralement comprise entre 20-80°C. Cette absence d’apport de
chaleur est très attractive pour le traitement de l’eau environnementale car il
n’est pas nécessaire de la refroidir après le traitement photocatalytique.
1.2.8-
Influence
du champ quantique
En photochimie, le concept appelé champ quantique est
utilisé pour évaluer les résultats obtenus et comparer différentes conditions
expérimentales. Le champ quantique (f) est définie par le rapport entre le nombre de molécules
qui ont réagi (Dn)
et la quantité de photons absorbés par le système (Na) :
![]() (57)
(57)
Le système hétérogène est constitué de TiO2
solide, d’oxygène gazeux (bulles ou dissous) et d’une multitudes de composés
aqueux (anions, protons, produits intermédiaires, …). En conclusion, la
quantité de photons absorbés par le catalyseur est très difficile à déterminer
expérimentalement. Pour la calculer, il faut :
- Evaluer
l’absorption de la lumière d’un mélange réactif très complexe qui,
toutefois, change sa composition par des réactions
- Déterminer
le flux de photons qui arrive à la surface du catalyseur pour le rendre
photoactif
- Estimer la
part de photons absorbés et dispersés
Ces travaux sont très difficiles à réaliser. Il faut se
souvenir que, en catalyse hétérogène, la vitesse de réaction est principalement
influencée par la quantité de catalyseur dans le système. En photocatalyse, il
faut inclure le nombre de sites actifs à la surface du catalyseur. Mais le
nombre de sites actifs ainsi que la surface de catalyseur irradiée sont indéterminés.
Afin de simplifier les calculs, l’irradiation du catalyseur
se fait à l’aide d’une source monochromatique. La valeur de Na
obtenue permet de calculer le champ quantique estimé fe. Aucune distinction n’est faite entre les photons
correspondant à chaque longueur d’onde, pourvu qu’ils aient tous le même effet
à la surface du catalyseur. Dans tout les cas, cette simplification est
acceptée comme valide par de nombreux auteurs. Par conséquent, les champs
quantiques rapportés ont été calculé sur une faible gamme de longueur d’onde
est par sur tout le spectre solaire.
La photo conversion du phénol a été choisie comme polluant
standard et le TiO2 DEGUSSA P25 comme photo conversion standard Ce compromis a
été adopté par un groupe de scientifique considéré comme les plus importants
dans le domaine. Le choix du phénol a été dicté par le fait que cette structure
moléculaire est présente dans presque tous les polluants organiques, et qu’il
est essentiellement dégradé par oxydation et non par réduction. On peut alors
introduire l’efficacité photonique relative (zr) par la relation suivante :
 (58)
(58)
Quand la vitesse de réaction pour les substances tests et
le phénol sont obtenues sous des conditions
expérimentales identiques, il n’est pas possible de mesurer le flux photonique
réellement efficace. L’utilisation de l’efficacité photonique relative permet
des comparaisons d’efficacité pour différents procédés réalisés aux
laboratoires et les installations semi-industrielles. L’efficacité photonique
relative est indépendante des paramètres photocatalytiques fondamentaux
(intensité lumineuse, géométrie du réacteur et concentration en TiO2).
Cependant, elle dépend de la concentration initiale du polluant et de la
température. Dans tout les cas, en se basant sur la cinétique de dégradation,
l’efficacité photonique relative illustre seulement un aspect de la
photodégradation et est utilisée pour
comparer différents systèmes photocatalytiques pour le traitement de l’eau.
1.2.9-
Influence
d’ions en solution
La présence d’ions métallique est courante dans les eaux
naturelles et dans les rejets industriels. Ces ions affectent sensiblement la
vitesse et l’efficacité des réactions photocatalytiques. Ainsi, la variation de
la vitesse de photodégradation a été observée pour les hydrocarbures
aromatiques, les acides benzoïques, les composés phénoliques, et d’autres
composés organiques en présence d’ions métallique, essentiellement Cu2+,
Fe3+ et Ag+ 31.
Sykora32 et coll. ont montré que l’influence des ions est fortement
dépendante du type et de la concentration de l’ion considéré. La variation de
la vitesse de photodégradation par ajout d’ions métalliques peut être
attribuée, dans un premier temps, à la faculté que possède cet ion à capturer
les électrons formés à la surface du catalyseur, réduisant ainsi la
recombinaison trou/électron. La vitesse d’annihilation trou/électron étant
réduite, la quantité de radicaux hydroxyles à la surface du TiO2 en
est augmentée.
Un des paramètres les plus important qui affecte
l’efficacité du processus photochimique est le potentiel rédox
standard du couple TiO2/Mn+
car seulement peut d’espèces avec un potentiel réducteur sont plus
positif que le bande de conduction et peuvent être photoréduit
(figure 5).
Figure 5 :
Les cations métalliques sont capables d’augmenter la
vitesse d’oxydation photocatalytique en participant également à des réactions
homogènes de type Fenton qui produisent également des radicaux hydroxyles OH°.
L’ion métallique peut réagir en l’absence de peroxyde d’hydrogène, mais la
vitesse d’oxydation peut augmenter encore plus par l’ajout de peroxyde
d’hydrogène (équation 15) :
![]() (59)
(59)
Si l’ion métallique est déposé à la surface du
semi-conducteur, les réactions de type Fenton n’ont pas lieu. Il a été montré
que les réactions de Fenton peuvent être réalisées en présence de lumière, on
parle alors de réaction de photo-Fenton. Dans les réactions classiques de
Fenton, la forme réduite de l’ion est consommée, et la réaction s’arrête
progressivement. L’avantage du système photo-Fenton est la régénération de la
forme réduite par oxydation photocatalytique :
![]() (60)
(60)
![]() (61)
(61)
L’effet de différents cations sur la dégradation
photocatalytique du phénol a été étudié par Brezova33 et coll.. Dans le cas de Ca2+, Mg2+, Ni2+,
Zn2+, Mn2+ et Co2+, la vitesse de dégradation
est fortement dépendante du potentiel réducteur du cation mais peu de la
concentration en ions. Les ions Ca2+, Mg2+, Zn2+
et Ni2+ ne montrent aucune influence sur la cinétique de
photodégradation du phénol. Les ions Mn2+, Co2+ et Cu2+
ralentie la dégradation du phénol. La présence d’ions Cr3+ arrête
totalement la réaction de dégradation.
L’effet des ions Mn2+ et Co2+ est
expliqué par un transfert d’électron entre la surface du catalyseur et les
ions. Cela entraîne une diminution de production de radicaux OH° et une
adsorption compétitive des ions avec le phénol. L’effet des ions Cr3+
est attribué au fait que les cations créés des sites accepteurs/donneurs
d’électron jouant le rôle de centre recombinaison.
Sykora32 et coll. ont montré que l’influence des
ions et fortement dépendant du type et de la concentration en ion considéré.
Ainsi, la photodégradation du phénol par TiO2 en suspension est
accélérée par la présence d’ions Cu2+ jusqu’à une concentration de 1
mmole ; au-delà, la cinétique de dégradation est
fortement diminuée.
L’effet néfaste d’une grande concentration en ions peut
s’expliquer par l’oxydation par les radicaux hydroxyles de l’ion métallique
réduit (équation 15). D’autres effets peuvent être cités. Sclafani34
et coll. ont étudié l’effet d’une grande concentration en ions Fe3+
sur la cinétique de photodégradation du toluène. Ils ont montré que les ions
absorbent les radiations U.V. nécessaire au photocatalyseur. De plus, les ions
métalliques précipitent sous forme d’hydroxyde et la solution devient de plus
en plus opaque au rayonnement35.
1.2.10- Influence de la méthode de préparation et de
la cristallinité du catalyseur
Des différences de vitesse de photodégradation ont été
remarquées pour des polluants organiques en solution aqueuse, en utilisant une
suspension de TiO2 préparé de différentes manières ou par la même
méthode mais dans des conditions expérimentales différentes. Il est possible de
comparer la photoactivité de différentes poudres et
d’expliquer pourquoi des catalyseurs, apparemment identiques par le point de
fusion, présentent des réactivités photocatalytiques différentes.
Les échantillons de TiO2 préparés, en général, à
partir de TiCl3 ou de TiCl4 par précipitation du
précurseur en milieu aqueux en utilisant de l’ammoniaque ou de la soude,
présentent des comportements catalytiques totalement différents pour la
photodégradation de phénol36.
Les échantillons dérivés de TiCl3 sont obtenus
principalement dans la phase anatase après un traitement thermique compris
entre 873-923 K pendant 3 à 192 heures. Ils présentent tous une activité
photocatalytique bien qu’à des degrés différents. La phase rutile peut être
obtenue après un traitement thermique plus long ou par une augmentation de la
température à 1073 K. Les phases obtenues ne sont photoactives
qu’en mélange avec de l’anatase.
Les échantillons dérivés de TiCl4 montrent une
quantité significative de rutile après un traitement thermique à l’air entre
673-823 K pendant 3 à 24 heures. Pour une température supérieure à 873 K et/ou
par une durée du traitement thermique plus longue, la phase rutile devient
unique ou dominante. Mais, plus significativement, en dehors d’une différence
structurale à cause des préparations différentes, les échantillons d’anatase
obtenus à partir de TiCl4 sont plus photoactifs
que ceux obtenus à partir de TiCl3.
Cependant, les échantillons obtenus à partir de TiCl4
et contenant principalement ou exclusivement de la phase rutile, sont plus photoactifs quand le traitement thermique n’excède pas 973
K pendant 3 heures. Une augmentation de la température et/ou de la durée du
traitement thermique, pour des échantillons préparés à partir de TiCl3,
forment une phase rutile très peu photoative.
Pour résumé les différents points :
-
des
différences de photoactivité sont mises en évidence
pour un même semi-conducteur (TiO2), pour la phase rutile et anatase
quand les méthodes de préparations sont différentes.
-
la phase
rutile obtenue à partir de TiCl4 est photoactive
quand elle est obtenue à une température inférieure à 973 K et inactive quand
la température est supérieure à 973 K.
-
la phase
rutile est obtenue à partir de TiCl3 seulement pour une température
de 1073 K ou après un long traitement thermique entre 923-973 K. Cette phase ne
présente pas de caractère photoactif mais quand elle est mélangée à de
l’anatase (obtenu par la même méthode mais à une température plus faible) la photoactivité est plus grande que celle des échantillons
préparés à partir de TiCl4.
La différence de photoactivité
est difficile à expliquer. Il a été montré que la vitesse de recombinaison
trou/électron sont significativement différentes pour
l’anatase et le rutile ; la vitesse est beaucoup plus grande pour le
rutile37. Ce paramètre joue un rôle néfaste sur la vitesse de
photodégradation des polluants car elle limite la formation des radicaux
hydroxyles nécessaires aux réactions.
Les facteur électroniques ne sont pas suffisant pour expliquer la différence de photoréactivité entre les deux phases, et plusieurs paramètres
physico-chimiques peuvent être considérés
-
la
surface hydroxylée du catalyseur est le plus important
car les groupements hydroxyles sont essentiels dans le mécanisme de
photodégradation. La présence de groupements hydroxyles à la surface du TiO2
favorise l’adsorption de O2 qui est réductible par capture
d’électron photoproduit. La faible réactivité
photocatalytique des échantillons préparés à hautes températures peut être
expliqué par une déshydratation irréversible de la surface du catalyseur,
diminuant la production de radicaux OH°.
-
La
taille des particules est un autre paramètre important. En effet, des petites
particules présentent une meilleure dispersion dans la phase aqueuse et
favorise donc les interactions photons/catalyseur/polluants à dégrader. De
petites particules permettent également une meilleure dispersion des photons
dans la solution à traiter.
-
La
surface spécifique du catalyseur est proportionnelle à la taille des
particules. Elle joue un rôle important dans les interaction
catalyseur/polluants. Il a été montré que plus la surface spécifique est
grande, plus les polluants peuvent s’adsorber à la surface du catalyseur et
peuvent réagir plus rapidement avec les radicaux hydroxyles formés à la surface
du TiO2 38. Plus la température du traitement thermique
est longue, plus la surface spécifique du catalyseur devient petite à cause
d’une déshydratation de la surface.
Pour une purification de l’eau, le photocatalyseur (TiO2)
peut être utilisé en suspension ou supporté39. Dans ce dernier cas,
le catalyseur est déposé sur une plaque de verre, de fibre de verre ou d’autres
types de support (tubes, …).
L’utilisation de catalyseur supporté dans l’oxydation
photochimique apporte un avantage majeur pour la séparation des particules catalytiques de l’eau traitée. Différents réacteurs à
catalyseur supporté ont été suggérés, et le seul problème majeur est de bien
fixer le film de catalyseur sur le support approprié.
Généralement, l’activité des différents catalyseurs est
comparée à la vitesse d’oxydation par unité de masse de photocatalyseur. Le TiO2
DEGUSSA P25 est utilisé comme standard de comparaison en suspension.
1.3-
Les réacteurs
photochimiques et les sources lumineuses
1.3.1-
Les
réacteurs photochimiques
a-
Réacteurs
de laboratoires
Pour les photoréacteurs, la géométrie et les relations
spatiales entre le réacteur et la source lumineuse sont très importantes. La
configuration géométrique du photoréacteur est déterminée pour obtenir un
maximum d’irradiation de la source lumineuse. L’irradiation peut être normale
ou parallèle à la surface du réacteur. En sélectionnant une configuration
géométrique, il est nécessaire de déterminer le chemin optique de la lumière
qui est obtenue dans le réacteur car c’est le facteur le plus important
affectant l’absorption lumineuse par le mélange réactionnel.
Les formes les plus courantes des photoréacteurs sont les
suivantes :
-
les
photoréacteurs à immersion : ce sont les réacteurs les plus simples
utilisés dans la majorité des laboratoires et des pilotes industriels. Il s’agit
d’un réservoir agité dans lequel les particules solides de catalyseur sont en
suspension dans l’eau. Une ou plusieurs lampes sont immergées dans la
suspension (figure 6). Ce système a l’avantage de pouvoir travailler en
continue. De plus, cette configuration
géométrique est très simple à mettre en place et permet d’obtenir une
efficacité photonique très grande. L’inconvénient majeur est qu’il est possible
de former un film de particules très fines à la surface de la lampe ce qui
augmente la dispersion de la radiation lumineuse.
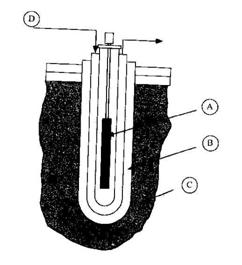
Figure 6 : photoréacteur à immersion ; A) lampe U.V., B)
isolation thermique, C) milieu réactionnel,
D)
eau de refroidissement
-
Les photoréacteur annulaires : la zone de réaction est
délimitée par deux cylindres coaxiaux. La lampe est placée dans l’axe de
symétrie. Ce système permet de travailler en continu et discontinu.
Pratiquement tous les photons émis par la lampe peuvent agir dans le milieu
réactionnel. Si l’épaisseur de la solution est faible, il est possible
d’ajouter un miroir autour du réacteur afin d’augmenter la quantité de photons
dans le milieu réactionnel (figure 7).
-
Les
photoréacteurs multi-lampes : le réacteur est
cylindrique, entouré de plusieurs lampes. Cette géométrie est souvent utilisée
lorsque les lampes sont fluorescentes car ont une puissance très faible.
Généralement, les surfaces réfléchissantes sont paraboliques et les lampes sont
placées au centre (figure 8).
-
Les
photoréacteurs elliptiques : le réacteur cylindrique et la lampe sont
placés au centre d’un cylindre réfléchissant elliptique. Dans cette géométrie,
la majeure partie des photons arrivent sur le réacteur
après réflexion sur la surface réfléchissante. Il a été montré que l’énergie
n’est pas uniforme dans le photoréacteur, et que l’intensité dépend des
paramètres de l’ellipse (figure 9).
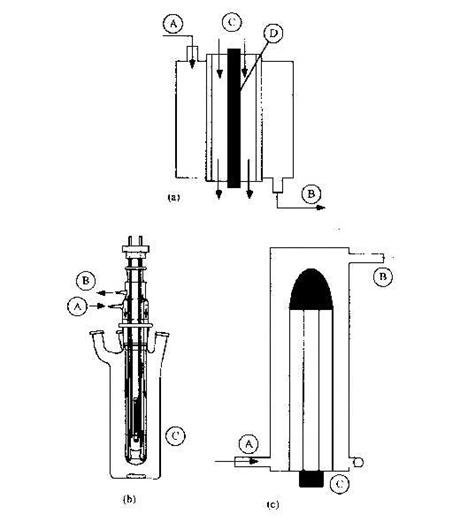
Figure 7 : photoréacteur
annulaire. (a) photoréacteur annulaire à débit constant : A) arrivé des
réactants, B) sortie des produits, C) liquide de refroidissement, D) lampe. (b)
photoréacteur annulaire : A) entrée de l’eau de refroidissement, B) sortie
de l’eau de refroidissement, C) lampe. (c) photoréacteur annulaire à lit
fluidisé : A) arrivé des réactant tangent à la lampe, B) sortie de l’eau
de refroidissement, C) lampe

Figure 8 : photoréacteur multilampe :
A) réacteur tubulaire, B) lampes, C) réflecteurs paraboliques

Figure
9 : photoréacteur élliptique. (a) vue du dessus.
(b) vue en perspective, A) réacteur cylyndrique, B) lampe, C) réflecteurs élliptiques,
D) propagation des photons
-
Les
photoréacteurs à film de catalyseur : le réacteur cylindrique est
constitué d’un film mince de catalyseur dans la paroi interne ou circule à la
solution. La lampe est placée au centre du réacteur (figure 10).

Figure 10 : photoréacteur à film de catalyseur. (a)
photoréacteur à irradiation extérieure (positive). (b) photoréacteur à
irradiation intérieure (négative). (c) photoréacteur à film mince
-
Les
photoréacteurs à plaques : le réacteur est constitué de deux plaques
parallèles, l’entrée et la sortie de la solution se font perpendiculaire au
sens de déplacement du liquide. L’irradiation lumineuse se fait face aux
plaques (figure 11).

Figure 11 : photoréacteur à plaques. (a) vue de face. (b) vue
de côté, 1) entrée de la solution,
2) sortie de la solution, 3) irradiation lumineuse
b-
Réacteurs
semi-industriels
Les différents collecteurs solaires ont été classé suivant le niveau de concentration obtenu à
l’intérieur. Le rapport de concentration peut être définie entre le rapport de
l’airre d’ouvrture du
collecteur qui absorbe les rayons solaires et l’aire totale du réacteur. L’aire
d’ouverture est l’aire interceptant les radiations et l’aire absorbante, l’aire recevant les
radiations solaires concentrées.
Trois catégories sont ainsi définies :
-
faible
concentration ou faible température : jusqu’à 150°C
-
moyenne
concentration ou température moyenne : entre 150 et 400°C
-
forte
concentration ou forte température : au delà de 400°C
Les collecteurs faibles concentrations sont statiques
(figure XX). Généralement, se sont des plaques plates orienté vers le soleil
suivant la localisation géographique. L’avantage d’un tel système est son
faible coût pour la mise en place.

Pour les collecteurs moyennes concentrations le rapport de
concentration est compris entre 5 et 50. De plus, ils nécessitent un système
qui suit le soleil durant la journée. Les collecteurs paraboliques et les
collecteurs holographiques ( collecteurs de Fresnel)
font parties de cette catégorie. Le premiers a une
surface de réflexion parabolique qui concentre les rayons solaires dans un
tubes au centre de la parabole. Le collecteur de Fresnel consiste à dévié les radiations solaires par des surfaces
réfléchissantes (similaire à des lentilles de Fresnel).

Les collecteurs fortes concentrations ont un point focal à l’intérieur
d’une ligne focale et sont placés sur une parabole suivant le soleil. Le
rapport de concentration est compris entre 100 et 10000 et une très grande
précision optique est nécessaire. Cette catégorie incluse les fours solaires.

1.3.2-
Les
sources lumineuses
a-
Irradiation
solaire
Le soleil est un énorme réacteur qui envoie en moyenne à la
surface de la Terre 1,5.1018 kWh par ans, ce qui représente
approximativement 28000 fois la consommation
mondiale annuelle. Les radiations hors de l’atmosphère ont une longueur
d’onde comprise entre 0,2 et 50 mm, qui est réduit entre 0,3 et 3 mm lorsqu’ils atteignent la surface à cause de l’absorption
par les différents composés atmosphériques (ozone, oxygène, dioxyde de
carbone,…). Les radiations qui atteignent la terre sans être absorbées ou
dispersées sont appelées radiations directes. Les radiations qui atteignent la
terre en étant dispersées sont appelées radiations diffuses, et la somme des
deux représente la radiation globale.
La figure XX représente les différents domaines de
rayonnement classés suivant les longueurs d’onde.

La photocatalyse hétérogène
repose sur l’irradiation d’un semi conducteur par un rayonnement UV. Le
rayonnement UV possède plus de propriétés quantiques que le rayonnement visible
ou infra-rouge. La lumière ultra-violette est généralement scindée en 3 parties
suivant les effets qu’elle produit.
La région UV-A (315-400 nm), qui
est le type de rayonnement le moins nuisible. On l’appelle souvent lumière
noire et est utilisé pour exciter des matériaux fluorescent
pour émettre une lumière visible, qui apparaît dans le noire.
La région UV-B (280-315 nm) est
typiquement la plus destructive, parce que l’énergie du rayonnement est
suffisante pour créer des dommages aux tissus biologiques. Ce domaine de
rayonnement est connu pour provoquer des cancers. Ce rayonnement peut être
complètement absorbé par l’atmosphère.
La région UV-C (100-280 nm) est
complètement absorbé par l’air. Quand les photons UV-C
entre en collision avec de l’oxygène de l’air, le transfert d’énergie est
suffisant pour former de l’ozone. Les lampes UV-C
sont utilisées pour le traitement de l’eau car le rayonnement permet de tuer
les bactéries.
b-
Irradiation
artificielle
La source de lumière est un facteur très important dans l’élaboration
de réacteur photochimique. Différentes lampes permettent la génération de
radiations pour différents domaines de longueurs d’onde. Le choix d’une lampe
se fait suivant l’énergie de réaction requis dans le
processus.
Il y a quatre types de source de radiations40 :
-
Les
lampes à arcs : l’émission est obtenue par un gaz activé par collisions
avec des électrons accélérés par une décharge électrique. Le gaz activé est en
général du mercure et/ou du xénon.
-
Les
lampes fluorescentes : l’émission est obtenue par l’excitation de
substances fluorescentes, déposées dans un cylindre, par décharge électrique
réalisée dans le gaz à l’intérieur du cylindre. Généralement, ces lampes
émettent dans la région visible, mais certaines lampes aux actinides ont une
émission dans le proche U.V. Il est évident que le spectre d’émission dépend de
la nature de la substance fluorescente utilisée. La puissance de ces lampes est
relativement faible, environ 150 W.
-
Les
lampes incandescentes : l’émission est obtenue par chauffage à très haute
température d’un filament, de nature variable, par circulation d’un courant
électrique.
-
Les
lasers : ils sont fréquemment utilisés en photochimie et dans bien
d’autres domaines. Ils produisent des radiations cohérentes et de très fortes
intensités.
En photocatalyse, les lampes à arcs ainsi que les lampes
fluorescentes sont fréquemment utilisées pour différentes raisons : en
utilisant le mercure ou le xénon, le spectre démission est très proche du
spectre solaire. De plus, ces lampes émettent peu de chaleur par rapport aux
autres lampes (incandescence et laser).
Références Bibliographiques
[1] BAUER R., WALDNER
G., MALATO S., The photo-fenton reaction and the
TiO2/UV process for waste water treatment – novel developments, Catalysis
Today, 53, 131-144, 1999.
[2] World Resources
Institute 1994-95, New York/Oxford, 1994.
[3] KOCH E, Global Trends 93/94,
Fischer Taschenbuchverlag, Frankfurt, 305, 1993.
[4] BONNEFONT-ROUSSELOT D.,
Oxydation des lipoprotéines et mécanisme d’action des anti-oxydants : apport de
la radiolyse gamma, Annales de Biologie Clinique, 57(4), 409-416, 1999.
[5] Spinks JWT, Woods
RJ. An introduction to radiation biology. Third Edition. John Wiley & Sons, Inc,
[6] Von Sonntag C. The chemical basis of radiation
biology.
[7] Fitchett
M, Gilbert BS, Wilison R.L. Fragmentation reactions
of radicals formed from sugar phosphates and the hydroxyl radical
: an investigation by electron spin resonance spectroscopy and pulse
radiolysis. J Chem Soc Perkin Transactions, 2, 673-89, 1988.
[8] Stadtman
ER. Oxidation of free amino acids and amino acid residues in
proteins by radiolysis and by metal-catalyzed reactions. Annu Rev Biochem, 62, 797-821,
1993.
[9] O'Neill P, Fielden EM. Primary free radical processes in DNA. Advances in radiation biology, vol. 17, 53-120. Academic
Press,
[10] Wallace
SS. Enzymatic processing of radiation-induced free radical damage in DNA. Radiat Res,
150, 60-79, 1998.
[11] MOSER J-E, Processus phoyochimiques, 2001.
[12] LE ROUX H., GLASSER
L., Transferable potentials for Ti-O system, Journal of Materials Chemistry,
7(5), 843-851, 1997.
[13] L. A. BURSILL, B.
G. HYDE, Prod. Solid
Chem., 7, 177, 1972.
[14] J. B. GOODENOUGH, Physical Review, 117, 1442, 1960.
[15] S. ANDERSON, A. D.
WADSLEY, Nature (
[16] D. T. CROMER, K.
HERRINGTON, Journal of American Chemical
Society, 77, 4708, 1955.
[17] E. P. MEAGHER, G.
A. LAGER,
[18]
P. SUPPAN, Chemistry and Light, Royal
Society of Chemistry,
[19] A. MILLS, S. LE
HUNTE, An overview of semiconductor photocatalysis, J. of Photochem. & Photobiol. A : Chem., 108, 1-35, 1997.
[20] A. FUJISHIMA, K.
HONDA, Nature, 238, 37, 1972.
[21] D. M. BLAKE, P. C.
MANESS, Z. HUANG, W.A. JACOBY, application of the photocatalytic
chemistry of TiO2 to disinfection and the killing of cancer cells, Separation and Purification Methods,
28 (1), 1-50, 1999.
[22]
K. RAJESHWAR, Photoelectrochemistry and the
environment; Journal of Applied
Electrochemistry, 25 (12), 1067-1082, 1995.
[23]
[24]
[25] J. CUNNINGHAM, P.
SEDLAK, Interrelationships between pollutant concentration, extent of
adsorption, TiO2 sensitized removal, photon flux and levels of
electron or hole traping additives, Journal of Photochemistry and
Photobiology A : Chemistry, 77,
255-263, 1994.
[26] V. BREZOVA, A.
BLAZKOVA, Phenol decomposition using Mn+/TiO2 photocatalysts supported by the sol-gel technic
on glass fibers, Journal
of Photochemistry and Photobiology A :
Chemistry, 109, 177-183, 1997.
[27]
[28] TOMIN
[29] I. BAUDIN, J. M.
LAINE, D. D. DIONYSIOU, M. T. SUIDAN, Effect of ionic strength and hydrogen
peroxide on the photocatalytic degradation of
4-chlorobenzoic acid in water, Applied
Catalysis B : Environmental, 26, 153-171, 2000.
[30] KORMANN
[31] M. I. LITTER,
Heterogeneous photocatalysis, transition metal ions
in photocatalytic systems, Applied Catalysis B : Environmental, 23,
89-114, 1999
[32]
J. SYKORA, Photochemistry of copper complexes and their environmental aspects, Coordination Chemistry Reviews, 159,
95-108, 1997.
[33] BREZOVA
[34] A. SCLAFANI, L.
PALMISANO, E. DAVI, Journal of
Photochemistry and photobiology A : Chemistry, 56,
113, 1991
[35] E. C.
[36] L. PALMISANO, A.
SCLAFANI, Thermodynamics and kinetics for heterogeneous photocatalytic
processes, Heterogeneous Photocatalysis, Wiley series in photoscience
and photoengineering, vol
3 (6), 109-132, 1997
[37] K. M. SCHINDLER, M.
KUNST, Journal of Physical Chemistry,
94, 8222, 1990
[38] H. TAMURA, N.
KATAYAMA, R. FURUICHI, Modeling of ion-exchange
reactions on metals oxides with the Frumkin isotherm,
Environmental Science and Technology,
30 (4), 1198-1204, 1996
[39] A. D. MODESTOV, O.
LEV, Photocatalytic oxidation of 2,4-dichlorophenoxyacetic
acetic with titania photocatalyst.
Comparaison of supported and suspended TiO2, Journal of Photochemistry and Photobiology
A : Chemistry, 112, 261-270, 1998
[40] V. AUGUGLIARO, V.
LODDO, M. SCHIAVELLO, Heterogeneous photocatalytic reactors : an assessment of fundamental engineering aspects,
Heterogeneous Photocatalysis, Wiley
series in photoscience and photoengineering,
vol 3 (6), 169-189, 1997